A main focus of our lab is to investigate the correlation between systemic metabolism and cancer incidence and progression, with the goal of identifying metabolic dependencies that could be targeted therapeutically in cancer patients.
Using models of lung, pancreatic and other tissue cancers, our lab aims at understanding:
Using models of lung, pancreatic and other tissue cancers, our lab aims at understanding:
- How tumors survive and thrive in nutrient-limiting environments
- How tumor growth and metabolism can be affected by the systemic metabolic state of the host (e.g. dietary restriction, obesity, insulin resistance)
- How the host systemic metabolic state can, itself, get affected by tumor growth and metabolism (e.g. cancer-associated cachexia, or energy-wasting syndrome)
Targeting polyamine synthesis in pancreatic cancer
Press release @Forbes: Pancreatic Cancer Breakthrough
Press release @Forbes: Pancreatic Cancer Breakthrough
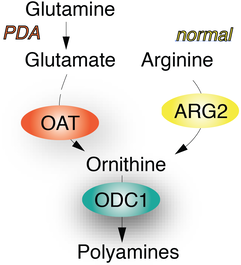
In our most recent work, we describe the identification of a unique metabolic dependency in pancreatic ductal adenocarcinoma (PDA), a highly lethal malignancy with no effective therapies. We find that PDA relies on de novo ornithine synthesis (DNS) from glutamine via ornithine aminotransferase (OAT), which supports polyamine synthesis and is required for tumor growth. This directional OAT activity is usually largely restricted to infancy and contrasts with the reliance of most adult normal tissues and other cancer types on arginine-derived ornithine for polyamine synthesis. We find that this dependence associates with arginine depletion in the PDA tumor microenvironment and is driven by mutant KRAS. Activated KRAS induces the expression of OAT and polyamine synthesis enzymes, leading to alterations in the transcriptome and open chromatin landscape in PDA tumour cells. The distinct dependence of PDA, but not normal tissue, on OAT-mediated de novo ornithine synthesis provides an attractive therapeutic window for treating patients with pancreatic cancer with minimal toxicity.
For more detail:
Check the News section on our lab website for News and Views, Commentaries and Press releases on this work
For more detail:
Check the News section on our lab website for News and Views, Commentaries and Press releases on this work
Halting KRAS-mutant lung cancer growth by starving it metabolically
Press release: Two-pronged approach could curb many cases of lung cancer
Press release: Two-pronged approach could curb many cases of lung cancer
To date, therapies are lacking that efficiently target the KRAS oncogene, which is responsible for approximately
a quarter of all lung cancer cases in the United
States. In this study, we provide genetic evidence that insulin / insulin-like growth factor 1 (IGF1) signaling, which modulates cellular survival, growth, and metabolism, is required for KRAS-driven lung cancer initiation.
We find that tumors with loss of insulin/IGF-1 signaling are metabolically starved, as they exhibit strikingly decreased nutrient (amino acid) uptake. As a result, they become highly dependent on autophagy (a self-eating process), and protein degradation, so as to compensate for decreased amino acid levels. Such metabolic vulnerability could be exploited therapeutically using clinically available autophagy and protein degradation inhibitors, in combination with insulin receptor/IGF1 receptor inhibitors, in patients with KRAS-mutant lung cancer.
a quarter of all lung cancer cases in the United
States. In this study, we provide genetic evidence that insulin / insulin-like growth factor 1 (IGF1) signaling, which modulates cellular survival, growth, and metabolism, is required for KRAS-driven lung cancer initiation.
We find that tumors with loss of insulin/IGF-1 signaling are metabolically starved, as they exhibit strikingly decreased nutrient (amino acid) uptake. As a result, they become highly dependent on autophagy (a self-eating process), and protein degradation, so as to compensate for decreased amino acid levels. Such metabolic vulnerability could be exploited therapeutically using clinically available autophagy and protein degradation inhibitors, in combination with insulin receptor/IGF1 receptor inhibitors, in patients with KRAS-mutant lung cancer.
In more detail:
Non–small-cell lung cancer (NSCLC) is a leading cause of cancer death worldwide, with 25% of cases harboring oncogenic KRAS. Although KRAS direct binding to and activation of PI3K is required for KRAS-driven lung tumorigenesis, the contribution of insulin receptor (IR) and insulin-like growth factor 1 receptor (IGF1R) in the context of mutant KRAS remains controversial.
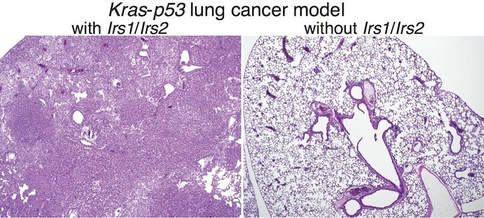
Here, we provide genetic evidence that lung-specific dual ablation of insulin receptor substrates 1/2 (Irs1/Irs2), which mediate insulin and IGF1 signaling, strongly suppresses tumor initiation and dramatically extends the survival of a mouse model of lung cancer with Kras activation and p53 loss. Mice with Irs1/Irs2 loss eventually succumb to tumor burden, with tumor cells displaying suppressed Akt activation and strikingly diminished intracellular levels of essential amino acids. Acute loss of IRS1/IRS2 or inhibition of IR/ IGF1R in KRAS-mutant human NSCLC cells decreases the uptake and lowers the intracellular levels of amino acids, while enhancing basal autophagy and sensitivity to autophagy and proteasome inhibitors.
These findings demonstrate that insulin/IGF1 signaling is required for KRAS-mutant lung cancer initiation, and identify decreased amino acid levels as a metabolic vulnerability in tumor cells with IR/IGF1R inhibition. Consequently, combinatorial targeting of IR/IGF1R with autophagy or proteasome inhibitors may represent an effective therapeutic strategy in KRAS-mutant NSCLC.
Non–small-cell lung cancer (NSCLC) is a leading cause of cancer death worldwide, with 25% of cases harboring oncogenic KRAS. Although KRAS direct binding to and activation of PI3K is required for KRAS-driven lung tumorigenesis, the contribution of insulin receptor (IR) and insulin-like growth factor 1 receptor (IGF1R) in the context of mutant KRAS remains controversial.
Here, we provide genetic evidence that lung-specific dual ablation of insulin receptor substrates 1/2 (Irs1/Irs2), which mediate insulin and IGF1 signaling, strongly suppresses tumor initiation and dramatically extends the survival of a mouse model of lung cancer with Kras activation and p53 loss. Mice with Irs1/Irs2 loss eventually succumb to tumor burden, with tumor cells displaying suppressed Akt activation and strikingly diminished intracellular levels of essential amino acids. Acute loss of IRS1/IRS2 or inhibition of IR/ IGF1R in KRAS-mutant human NSCLC cells decreases the uptake and lowers the intracellular levels of amino acids, while enhancing basal autophagy and sensitivity to autophagy and proteasome inhibitors.
These findings demonstrate that insulin/IGF1 signaling is required for KRAS-mutant lung cancer initiation, and identify decreased amino acid levels as a metabolic vulnerability in tumor cells with IR/IGF1R inhibition. Consequently, combinatorial targeting of IR/IGF1R with autophagy or proteasome inhibitors may represent an effective therapeutic strategy in KRAS-mutant NSCLC.
Nitrogen metabolism is a dependency in pancreatic cancer
Press release: A metabolic treatment for pancreatic cancer?
Press release: A metabolic treatment for pancreatic cancer?

In this work, we identify a novel metabolic dependency in obesity-associated pancreatic ductal adenocarcinoma (PDAC), the most common form of pancreatic cancer. PDAC is a highly lethal malignancy on the rise, characterized by extremely low (8-9%) 5-year survival rates.
We find that enhanced in vivo PDAC growth under an obese state results in the induction of enzymes, particularly arginase 2 (ARG2), that are involved in the disposal of excess nitrogen, a byproduct of protein and amino acid degradation.
Interestingly, this dependency is not restricted to the obese, as PDAC tumors carrying oncogenic
mutations (e.g. AKT) that enhance in vivo growth also induce and are dependent on ARG2
expression.
We find that enhanced in vivo PDAC growth under an obese state results in the induction of enzymes, particularly arginase 2 (ARG2), that are involved in the disposal of excess nitrogen, a byproduct of protein and amino acid degradation.
Interestingly, this dependency is not restricted to the obese, as PDAC tumors carrying oncogenic
mutations (e.g. AKT) that enhance in vivo growth also induce and are dependent on ARG2
expression.
In more detail:
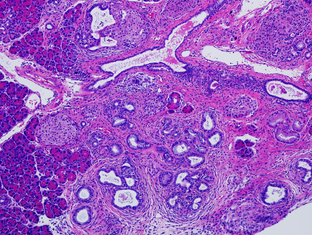
Obesity is an established risk factor for PDAC. Despite recent identification of metabolic alterations in this lethal malignancy, the metabolic dependencies of obesity-associated PDAC remain unknown. We show that obesity-driven PDAC exhibits accelerated growth and a striking transcriptional enrichment for pathways regulating nitrogen metabolism. Specifically, we find that the mitochondrial, extrahepatic form of arginase (ARG2), which hydrolyzes arginine into ornithine and urea, is induced upon obesity. In vivo infusion of 15N-glutamine in obese mouse models of human PDAC demonstrates that this induction is accompanied by enhanced nitrogen flux into the urea cycle, the major metabolic pathway for ammonia detoxification, normally operating in mammalian liver. Silencing or loss of ARG2 markedly suppresses PDAC, resulting in significant ammonia accumulation and the shunting of arginine catabolism into alternative nitrogen repositories.
Interestingly, this dependency is not restricted to the obese, as PDAC tumors carrying oncogenic
mutations (e.g. AKT) that enhance in vivo growth in the lean also induce and are dependent on ARG2 expression. Furthermore, analysis of resected tumors from PDAC patients indicates that ARG2 levels correlate with body mass index (BMI) and with activated AKT, independent of BMI.
It is noteworthy that unlike its hepatic cytosolic isoform ARG1, deficiencies in which result in hyperammonemia, seizures and even death, no human diseases are associated with ARG2. The specific dependency of PDAC on ARG2 rather ARG1 opens a therapeutic window for the treatment of pancreatic cancer patients suffering from obesity, as well as lean patients carrying tumors with AKT-activating mutations, independent of obesity.
Obesity is an established risk factor for PDAC. Despite recent identification of metabolic alterations in this lethal malignancy, the metabolic dependencies of obesity-associated PDAC remain unknown. We show that obesity-driven PDAC exhibits accelerated growth and a striking transcriptional enrichment for pathways regulating nitrogen metabolism. Specifically, we find that the mitochondrial, extrahepatic form of arginase (ARG2), which hydrolyzes arginine into ornithine and urea, is induced upon obesity. In vivo infusion of 15N-glutamine in obese mouse models of human PDAC demonstrates that this induction is accompanied by enhanced nitrogen flux into the urea cycle, the major metabolic pathway for ammonia detoxification, normally operating in mammalian liver. Silencing or loss of ARG2 markedly suppresses PDAC, resulting in significant ammonia accumulation and the shunting of arginine catabolism into alternative nitrogen repositories.
Interestingly, this dependency is not restricted to the obese, as PDAC tumors carrying oncogenic
mutations (e.g. AKT) that enhance in vivo growth in the lean also induce and are dependent on ARG2 expression. Furthermore, analysis of resected tumors from PDAC patients indicates that ARG2 levels correlate with body mass index (BMI) and with activated AKT, independent of BMI.
It is noteworthy that unlike its hepatic cytosolic isoform ARG1, deficiencies in which result in hyperammonemia, seizures and even death, no human diseases are associated with ARG2. The specific dependency of PDAC on ARG2 rather ARG1 opens a therapeutic window for the treatment of pancreatic cancer patients suffering from obesity, as well as lean patients carrying tumors with AKT-activating mutations, independent of obesity.
Starved, normal epithelial cells uptake extracellular matrix for survival
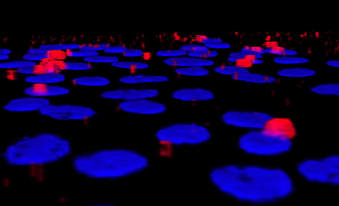
In a collaboration with the Brugge and Muranen labs, we identified a previously unrecognized mechanism for maintenance of epithelial cell homeostasis under conditions of nutrient starvation. Unexpectedly, it involves the uptake by the epithelial cells of extracellular matrix for survival.
In more detail:
We find that under conditions of dietary restriction in vivo or growth factor starvation and decreased PI3K/mTOR signaling in vitro, which mimics loss of nutrient uptake, normal epithelial cells utilize an alternative, unconventional means to maintain nutrient supplies. Unexpectedly, this involves uptake of extracellular matrix, specifically laminin, which upon binding b4-integrin, is endocytosed and internalized into the lysosomes. This in turn leads to increased intracellular amino acid levels, re-activating mTORC1 and promoting survival. The use of laminin as a nutrient source in epithelial cells with suppressed mTORC1 signaling would allow the cells to survive transient periods of nutrient starvation, leading to re-activation of mTORC1 and induction of pro-survival signaling. Such periods of starvation mimic those experienced by hypovascular tumors growing in vivo.
Moreover, we describe a crosstalk between human fibroblasts and mammary epithelial cells, where laminin secretion by starved fibroblasts enhances mTORC1 signaling and survival of starved epithelial cells, contingent upon the epithelial cell b4-integrin expression. These findings provide a potential therapeutic opportunity through targeting of integrins in stromally-enriched cancers that are resistant to PI3K/mTOR inhibition, which mimics starvation.
In more detail:
We find that under conditions of dietary restriction in vivo or growth factor starvation and decreased PI3K/mTOR signaling in vitro, which mimics loss of nutrient uptake, normal epithelial cells utilize an alternative, unconventional means to maintain nutrient supplies. Unexpectedly, this involves uptake of extracellular matrix, specifically laminin, which upon binding b4-integrin, is endocytosed and internalized into the lysosomes. This in turn leads to increased intracellular amino acid levels, re-activating mTORC1 and promoting survival. The use of laminin as a nutrient source in epithelial cells with suppressed mTORC1 signaling would allow the cells to survive transient periods of nutrient starvation, leading to re-activation of mTORC1 and induction of pro-survival signaling. Such periods of starvation mimic those experienced by hypovascular tumors growing in vivo.
Moreover, we describe a crosstalk between human fibroblasts and mammary epithelial cells, where laminin secretion by starved fibroblasts enhances mTORC1 signaling and survival of starved epithelial cells, contingent upon the epithelial cell b4-integrin expression. These findings provide a potential therapeutic opportunity through targeting of integrins in stromally-enriched cancers that are resistant to PI3K/mTOR inhibition, which mimics starvation.
What modulates tumor sensitivity to dietary restriction?
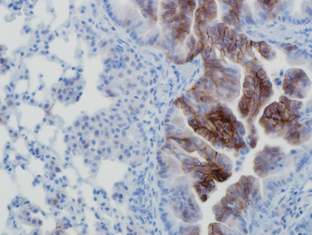
Dietary restriction (DR) has long been recognized for its anti-tumorigenic effects. However, the mechanisms underlying this effect have remained largely unknown. Our previous work showed that not all tumors are equally DR-sensitive and that tumors with PI3K/AKT activation are resistant to DR. Activation of this frequently mutated signaling pathway could result from either oncogenic PI3K mutations or loss of the PI3K antagonist and tumor suppressor PTEN. Indeed, PTEN loss has long been considered a biomarker for activated PI3K/AKT.
In this study, we aimed at investigating whether genetic loss of PTEN is an accurate indicator of the resistance of tumors DR. We show that PTEN loss is not sufficient to drive AKT activation and resistance to DR in tumors with low growth factor receptor levels. We describe a murine PTEN-null Kras-driven lung cancer model that harbors both DR-resistant, higher-grade, bronchiolar tumors with high AKT activity, and DR-sensitive, lower-grade, alveolar tumors with low AKT activity. We find that this phenotype is cell autonomous and that normal bronchiolar cells express higher levels of insulin-like growth factor-1 receptor (IGF-1R) and of ectonucleoside triphosphate diphosphohydrolase 5 (ENTPD5), an endoplasmic reticulum enzyme known to modulate growth factor receptor levels. Suppression of ENTPD5 is sufficient to decrease IGF-1R levels and sensitize bronchiolar tumor cells to serum in vitro and to DR in vivo. Furthermore, we find that a significant percentage of human non-small cell lung carcinomas have low AKT activity despite PTEN loss.
These results point to a heterogeneity of AKT activation in PTEN-null tumors further underscoring the challenges of personalizing cancer therapy based solely on cancer genotype. Moreover, our findings indicate that the tumor response to anticancer therapies, including DR, needs to be based on PI3K/AKT activity per se, rather than on genetic alterations in the PTEN/PI3K pathway.
In this study, we aimed at investigating whether genetic loss of PTEN is an accurate indicator of the resistance of tumors DR. We show that PTEN loss is not sufficient to drive AKT activation and resistance to DR in tumors with low growth factor receptor levels. We describe a murine PTEN-null Kras-driven lung cancer model that harbors both DR-resistant, higher-grade, bronchiolar tumors with high AKT activity, and DR-sensitive, lower-grade, alveolar tumors with low AKT activity. We find that this phenotype is cell autonomous and that normal bronchiolar cells express higher levels of insulin-like growth factor-1 receptor (IGF-1R) and of ectonucleoside triphosphate diphosphohydrolase 5 (ENTPD5), an endoplasmic reticulum enzyme known to modulate growth factor receptor levels. Suppression of ENTPD5 is sufficient to decrease IGF-1R levels and sensitize bronchiolar tumor cells to serum in vitro and to DR in vivo. Furthermore, we find that a significant percentage of human non-small cell lung carcinomas have low AKT activity despite PTEN loss.
These results point to a heterogeneity of AKT activation in PTEN-null tumors further underscoring the challenges of personalizing cancer therapy based solely on cancer genotype. Moreover, our findings indicate that the tumor response to anticancer therapies, including DR, needs to be based on PI3K/AKT activity per se, rather than on genetic alterations in the PTEN/PI3K pathway.